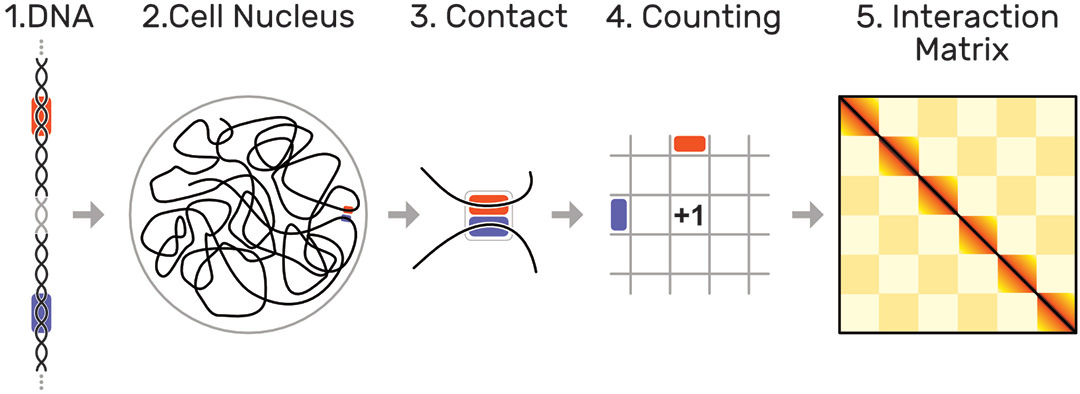
The human genome is about 2 meters long and tightly folded into each cell nucleus. This results in a dense, fractal-like and three-dimensional structure in which genome sequences that are distant on the genome, can be in close spatial proximity. It has been shown [1] that this 3D structure is an important factor for regulation of gene expression, replication, DNA repair, and other biological functions. Biologists are interested in uncovering the mechanisms that drive global and local folding to better understand the vast and complex gene regulation network.
The probability of two sequences being in close proximity to each other, i.e. interacting, can be inferred using modern genome sequencing techniques, which yield for every genome a huge symmetric genome interaction matrix with up to 3 million rows and 3 million columns (Fig. 1). Each of the 9 trillion matrix cells represents the proximity of two genomic regions. Repetitive and hierarchically nested visual patterns (Fig. 2), ranging from millions down to a few thousand base pairs in size, can be identified across the matrix, which represent so called regions of interest (ROIs).
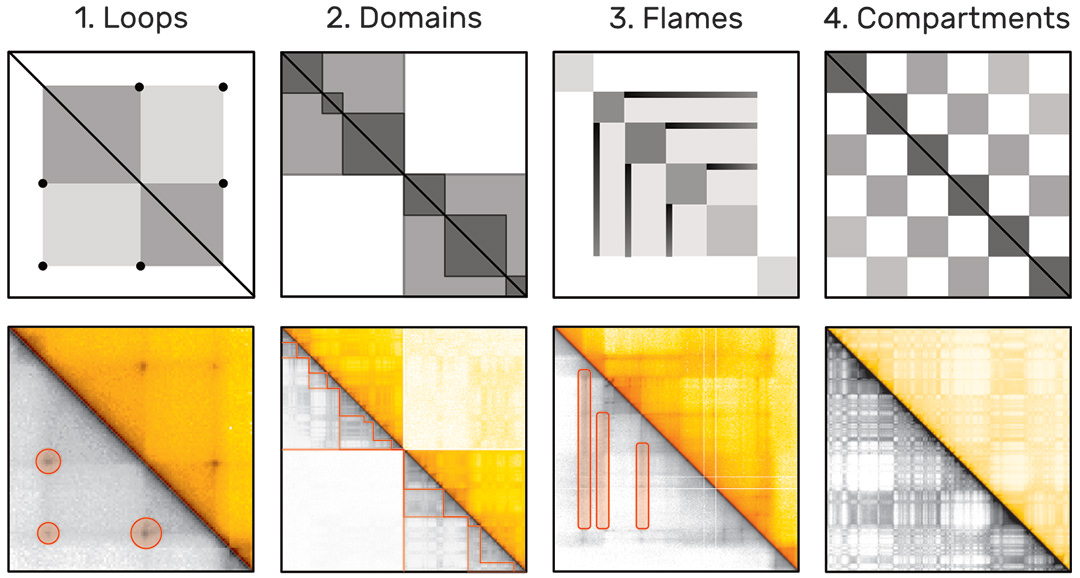
Algorithms for automatic pattern detection are being development. However, these algorithms can be very complex and often identify tens of thousands of pattern instances. Results of algorithms designed to identify the same type of pattern often differ substantially [2] and the lack of a ground-truth pattern collection hinders thorough evaluation of these algorithms.
Interactive visualization tools have been developed [3] but are focused on supporting visualization of a single or a small number of views of the matrix and navigation through pan and zoom [4, 5]. Detailed exploration and comparison of thousands of small ROIs is unsupported by current tools yet needed, due to the size and multiscale nature of the folded genome.
HiPiler is an interactive visualization tool designed for exploration and analysis of thousands of ROIs extracted from one or more genome interaction matrices.
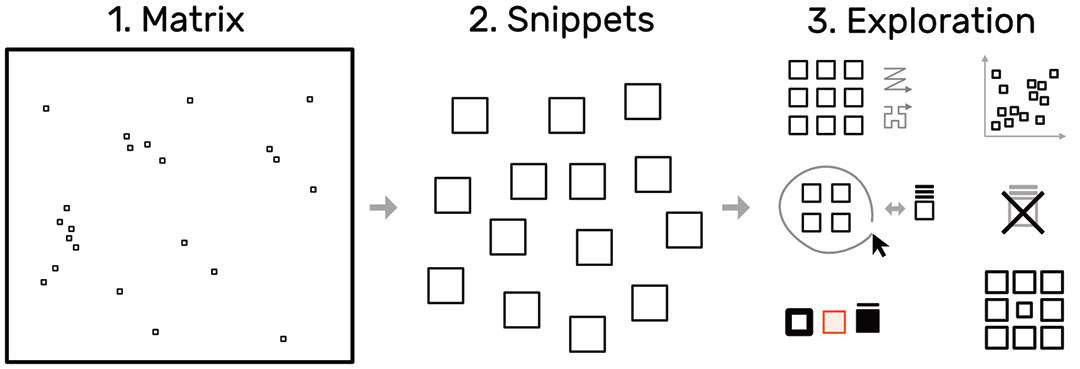
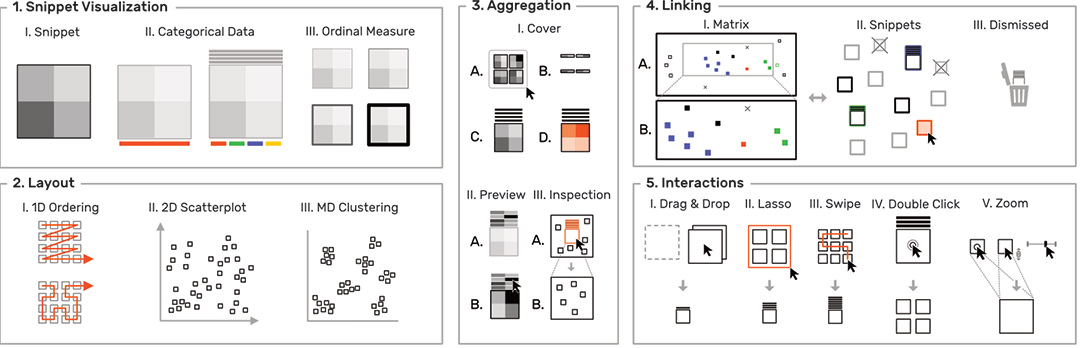
To overcome the contextual constraints of exploring local patterns in large matrices, HiPiler follows a divide and explore approach that extracts ROIs from the matrix and enables independent exploration (Fig. 3). HiPiler assumes a given set of ROIs, derived from specialized pattern recognition algorithms. HiPiler then visualizes these ROIs as small heatmaps (matrices) which we call snippets. A snippet is associated with a set of ordinal and categorical attributes, such as its noisiness, size, or source dataset. This data is derived from the matrix itself or point to prior knowledge. Based on this data, HiPiler enables automatic and manual ordering, positioning, grouping, filtering, and visual manipulation to identify patterns present across the set of snippets. Additionally, the context of snippets in the matrix is maintained through highlighting of snippet locations in the interaction matrix.
The design of HiPiler (Fig. 4) is informed by semi-structured interviews with ten domain experts from various genomics research labs as well as iterative design sessions over the course of several months. These interviews led to the formulation of six tasks for the exploration of many ROIs in large matrices.
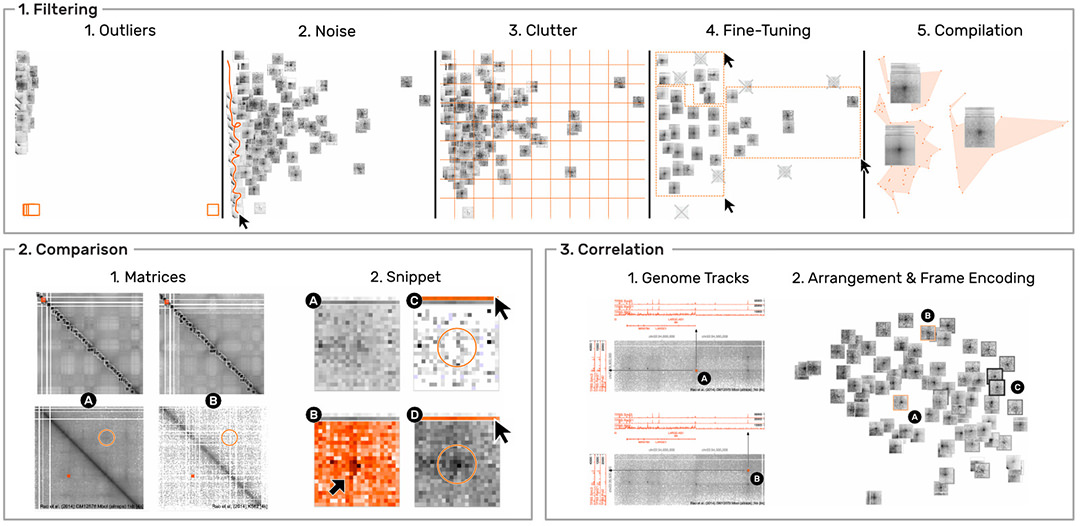
HiPiler is designed to support four types of scenarios. (i) visual evaluation of the results of pattern detection algorithms. (ii) characterization, aggregation, and outlier detection in large pattern collections (Fig. 5.1). (iii) comparison of ROIs across multiple matrices (Fig. 5.2), e.g., to compare different datasets, experimental conditions, or extraction algorithms.
And (iv) correlation of matrix patterns with other genomic attributes (Fig. 5.3), e.g., genes or protein-binding sites. We evaluated the usability and appropriateness of HiPiler through a user study with five domain experts. The results show that HiPiler is easy to learn and use, and that it offers important benefits for analyzing genome interaction matrices.